- LANTHANE ET LANTHANIDES
- LANTHANE ET LANTHANIDESLe lanthane et les lanthanides forment une série de quinze éléments métalliques de propriétés chimiques très semblables, que l’on désigne aussi plus communément sous le nom, d’ailleurs impropre, de «terres rares» (parce qu’on les a d’abord extraits à l’état d’oxydes ressemblant aux alcalino-terreux, à partir de minéraux peu courants). Dans la classification périodique des éléments, les lanthanides occupent, avec le lanthane, une seule et même case du tableau; cette particularité résulte de leur structure électronique, qui est identique pour les couches extérieures et ne diffère d’un élément au suivant que par addition d’un électron dans la couche profonde 4f (d’où le nom d’éléments de transition 4f que leur donnent parfois les physiciens).La liste des lanthanides et celle de leurs principales caractéristiques atomiques sont données dans le tableau 1. L’élément 61, le prométhéum (ou prométhium), n’existe pas dans la nature, mais c’est un composant assez abondant des produits de fission de l’uranium. Tous les autres lanthanides se trouvent toujours associés dans les minéraux, bien qu’en proportions assez variables, les éléments pairs étant en général plus abondants que les éléments impairs. Ils sont, en outre, toujours associés à l’yttrium, leur homologue situé au-dessus d’eux dans la colonne III A du tableau périodique [cf. ATOME].La prédominance dans les minéraux, soit des lanthanides légers, soit de l’yttrium et des lanthanides lourds, de même que l’évolution des propriétés chimiques et de la structure électronique, avec une certaine discontinuité au centre de la série, ont conduit à subdiviser la famille en un «groupe cérique» (du lanthane au gadolinium) et un «groupe yttrique» (du gadolinium au lutétium). Néanmoins, la similitude des propriétés chimiques des lanthanides (trivalence uniforme en solution) a constitué longtemps l’obstacle majeur au développement de leur étude et de leur utilisation industrielle. À l’exception de quelques éléments pouvant présenter des valences anormales, principalement le cérium, facilement oxydable à la valence + 4, les lanthanides sont restés, jusque vers les années quarante, des curiosités de laboratoire et n’étaient obtenus à l’état relativement pur que par des séparations chimiques extrêmement longues et laborieuses (cristallisations ou précipitations fractionnées). C’est seulement avec le développement de l’industrie nucléaire et la mise au point de nouvelles techniques de séparation (échanges d’ions, extraction par solvants) qui lui étaient nécessaires que les composés des lanthanides sont devenus des produits chimiques plus courants. Leur étude s’est alors considérablement développée, spécialement dans le domaine de la recherche fondamentale où ces éléments offrent un champ d’investigation pratiquement unique pour la vérification des théories de la physique du solide. Des confirmations spectaculaires ont alors été obtenues, de nouvelles propriétés découvertes et étudiées; les lanthanides sont alors considérés comme des matériaux industriels importants par la spécificité de leurs caractéristiques, d’autant plus que leurs minerais se sont révélés beaucoup plus abondants qu’on ne le supposait autrefois.1. État naturelLes lanthanides, contrairement à leur dénomination de «terres rares», sont des éléments assez répandus: leur concentration globale dans la croûte terrestre est de l’ordre de 0,016 p. 100, c’est-à-dire aussi élevée que celle du zinc, dix fois plus que celle du plomb, mille fois plus que celle de l’argent; l’élément le plus abondant de la famille (environ le tiers), le cérium, se situe entre le cuivre et l’étain, les deux plus rares, le thulium et le lutétium, entre le mercure et le cadmium. Mais, en raison de leur grande dispersion à la surface du globe, leur relative abondance n’a été mise en évidence que progressivement, au fur et à mesure que les méthodes de détection et d’analyse se sont perfectionnées, et que la recherche de leurs minerais s’est développée. Du point de vue géochimique, les lanthanides appartiennent au groupe des éléments «lithophiles», mais, au cours de la solidification du magma des silicates, ils se sont séparés des éléments communs du groupe, en raison du grand volume de leurs ions, et ont cristallisé finalement en phases distinctes avec d’autres éléments rares (Ti, Nb, Ta, Th). Il en résulte que les lanthanides sont le plus souvent disséminés en inclusions dans les grandes masses de roches ignées, de préférence dans les pegmatites, granites ou syénites néphéliniques, et certaines de leurs principales zones de concentration naturelle dérivent de la destruction progressive de ces roches anciennes.Les minéraux de lanthanides sont très nombreux. On en a décrit plus de deux cents, de compositions très diverses, avec prédominance, soit du groupe cérique, soit du groupe yttrique. Parmi les principaux, citons les carbonates et fluocarbonates (lanthanite, bastnaésite, parisite), les phosphates (monazite et xénotime), les silicates (cérite, allanite, gadolinite, thalénite), les niobo-tantalates ou titanates (samarskite, fergusonite, euxénite, polycrase, loparite). Mais deux d’entre eux seulement forment des gisements importants exploités industriellement: la monazite , à la fois minerai de terres rares et de thorium, que l’on rencontre en dépôts de sables alluvionnaires ou en masses cristallines (au Brésil, en Inde, aux États-Unis, en Afrique du Sud, Australie, Malaisie, Corée, Indonésie), et surtout la bastnaésite , dont il existe en Californie un gisement qui constitue sans doute la plus grande réserve naturelle de lanthanides du monde (estimée à 3 millions de tonnes d’oxydes); on en trouve également en Suède, au Nouveau Mexique, en Malaisie et au Congo; ces deux minerais contiennent essentiellement des éléments cériques. Les éléments du groupe yttrique (lanthanides lourds) sont extraits, en quantités beaucoup plus modestes, de la gadolinite (Texas et Idaho, Scandinavie, Europe centrale), du xénotime (Australie, Malaisie, Nigeria, Scandinavie, Brésil), de l’euxénite et de la samarskite (Scandinavie, U.R.S.S., États-Unis, Inde).Les lanthanides se trouvent aussi en quantités non négligeables dans des minéraux d’autres éléments, dans lesquels ils s’introduisent par substitution, remplaçant, en particulier, le calcium (dont le rayon ionique est proche de ceux des éléments cériques); la compensation des charges de valence s’opère par formation de lacunes par échange d’un autre cation (par exemple Nb5+ par Ti4+, Si4+ par Al3+, Al3+ par Mg2+) ou par insertion de radicaux anioniques dans le cristal (par exemple - ou OH-). Des minéraux tels que les fluorures (yttrofluorite), les phosphates (apatites, pyromorphite), les tungstates (scheelite), les uranates (uraninite, davidite), les silicates (orthite) et silico-titanates (titanites) contiennent ainsi souvent des proportions appréciables de lanthanides, qui peuvent être extraits avantageusement au cours du traitement de ces minerais (c’est le cas des apatites traitées pour la production d’acide phosphorique au Canada, aux États-Unis et en ex-U.R.S.S.). Il en est de même des minerais contenant des minéraux de lanthanides en inclusions, tels les minerais d’uranium du Canada et d’Afrique du Sud et les gisements de cassitérite de Malaisie.La production annuelle mondiale de lanthanides, exprimée en oxydes, s’est élevée, en 1988, à environ 55 000 tonnes, provenant de la bastnaésite et de la monazite venant des États-Unis et de la Chine. Le surplus a été fourni par la mise sur le marché d’une partie des stocks de l’industrie nucléaire américaine. La France possède en Bretagne un gisement de monazite riche en europium, mais inexploitable vu le prix de revient de l’extraction.2. Historique: une longue querelleL’histoire de la découverte des lanthanides est une des plus longues et des plus compliquées de toutes celles qui concernent les sciences physico-chimiques. Le premier minerai de lanthanides (cérite) fut trouvé en 1750, mais considéré d’abord comme un composé de tungstène: il fallut plus de cinquante ans pour que, simultanément, Martin Heinrich Klaproth et Jöns Jacob Berzelius y mettent en évidence un oxyde encore inconnu, la céria. Entre-temps, Johan Gadolin avait découvert en 1794, dans un minerai suédois, la première «terre rare», dénommée d’abord «ytterbia», puis «yttria». L’étude de la céria et de l’yttria montra par la suite qu’en réalité ces substances étaient des mélanges complexes de plusieurs éléments inconnus, mais chimiquement identiques. Des recherches laborieuses et acharnées, auxquelles participèrent de très nombreux chimistes et spectroscopistes, permirent, après bien des erreurs et de fausses découvertes, de dégager progressivement la liste de ces éléments: dans le groupe cérique, le lanthane et le cérium (Carl Gustaf Mosander, 1841), puis le samarium (Paul Émile Lecoq de Boisbaudran, 1879), le néodyme et le praséodyme (Carl Auer von Welsbach, 1885), ceux-ci considérés pendant plus de quarante ans comme un élément unique, le «didyme» de Mosander; enfin l’europium (William Crookes ou Eugène Demarçay, 1890). L’élément 61, le prométhium, dont Henry Moseley avait clairement démontré l’existence théorique dans la classification périodique, fut plusieurs fois à tort «découvert» et baptisé (illinium, florentium) avant qu’il ne soit enfin isolé comme élément radioactif dans les produits de fission de l’uranium (J. A. Marinsky, L. E. Glendenin et C. D. Coryell, 1947). Dans le groupe yttrique, c’est également aux travaux de Mosander que l’on doit la distinction entre l’yttrium et les lanthanides lourds, et la découverte du terbium et de l’erbium (1835); furent identifiés ensuite le gadolinium et l’ytterbium (Jean Charles Gallissard de Marignac, 1878), l’holmium et le thulium (Per Teodor Cleve, 1879), le dysprosium (Lecoq de Boisbaudran, 1886) et finalement le lutétium (Georges Urbain, 1907). En plus de l’yttrium, plusieurs éléments n’appartenant pas au groupe des lanthanides furent découverts au cours des travaux d’identification de ceux-ci: le scandium (l’homologue le plus léger de la colonne III A), le thorium, le béryllium et le hafnium.Pendant la première moitié du XXe siècle, les chimistes poursuivirent avec acharnement leurs efforts pour séparer les différents éléments du groupe; les seuls procédés alors connus étaient les précipitations ou les cristallisations fractionnées, et des dizaines de milliers d’opérations étaient nécessaires dans certains cas pour atteindre un degré de pureté suffisant. C’est néanmoins au cours de cette période que furent découvertes les principales propriétés des lanthanides, grâce en particulier aux travaux de savants français: propriétés de luminescence (Urbain) et propriétés magnétiques (Félix Trombe). Parallèlement, les lanthanides connaissaient un certain développement industriel, mais leur utilisation restait limitée à leurs mélanges (mischmétal pour alliages pyrophoriques, fluorures pour charbons à arc) et aux oxydes de cérium et de lanthane, faciles à séparer.Une période tout à fait différente, caractérisée par un développement d’une rapidité exceptionnelle, du point de vue aussi bien de la recherche que des applications, débuta à la fin de la Seconde Guerre mondiale, après que, sous la pression des nécessités de l’industrie nucléaire, on eut mis au point de nouvelles méthodes de séparations permettant la production à grande échelle de tous les lanthanides purs. De très nombreux composés nouveaux furent alors préparés et étudiés systématiquement, et les lanthanides sont devenus en quelques années des éléments importants par l’originalité de leurs propriétés et les applications spécifiques que l’on a pu en tirer dans les techniques de pointe (électronique, télévision, magnétisme, catalyse).3. ÉlaborationPartant de matières premières de composition généralement très complexe, les opérations qui permettent d’obtenir les différents lanthanides sous forme de produits purs sont longues et délicates. On peut y distinguer trois étapes successives: le traitement des minerais, l’isolement du groupe des terres rares, la séparation des éléments du groupe.Traitement des mineraisLes minerais de lanthanides étant de nature et de composition très diverses, il n’existe pas de méthode commune pour leur traitement. Industriellement, les opérations commencent toujours par une concentration mécanique: le minerai est broyé, lavé et criblé sur des tables à secousses ou sur des appareils rotatifs, généralement sur les lieux mêmes de l’extraction; on le transporte ensuite dans les ateliers de concentration. Dans le cas des minerais de monazite, dont la teneur est faible (de 0,1 à 5 p. 100), et qui renferment de fortes proportions de silice et d’autres minéraux (rutile, ilménite, zircon), des séparateurs électromagnétiques ou électrostatiques et des appareils à triage gravimétrique en courant d’air sont mis en œuvre pour obtenir finalement un concentré à plus de 99 p. 100. On utilise aussi parfois les procédés de flottation, mais cette technique est surtout appliquée au traitement de la bastnaésite: le minerai brut, assez riche (10 p. 100 d’oxydes de lanthanides en moyenne), est pulvérisé et trié dans l’eau (sur classeur de Dorr, par exemple), puis la bastnaésite est séparée de la majeure partie de la barytine et des autres impuretés qui l’accompagnent (silice, calcite, fluorine), par une flottation en émulsion à chaud. À partir de ce concentré (65 p. 100), un lavage à l’acide chlorhydrique dilué et une calcination donnent par décomposition du fluorocarbonate un oxyde brut titrant plus de 90 p. 100 d’oxydes de lanthanides.Les autres minerais de terres rares se traitent par des procédés analogues, faisant intervenir suivant les cas les techniques de triage hydraulique, les séparations électromagnétiques ou la flottation. Quant aux lanthanides contenus dans les minerais d’autres éléments, leur récupération n’est réalisée industriellement que si elle peut s’insérer dans le circuit normal de traitement de ces minerais, ou, plus tard, au cours des opérations chimiques d’extraction.L’attaque chimique des minerais peut se faire soit par un acide fort (sulfurique, chlorhydrique ou nitrique), soit par une solution alcaline (soude ou carbonate). Ces deux modes d’attaque conviennent bien pour les fluocarbonates (bastnaésite, yttroparisite), les phosphates (monazite, xénotime) et les silicates; l’acide sulfurique est utilisé aussi pour les minerais titanifères (yttrotitanite, euxénite, polycrase). Pour décomposer les minerais difficiles à attaquer, en particulier ceux qui contiennent du titane, du niobium ou du tantale, on a recours à l’acide fluorhydrique, et, surtout au laboratoire, à des fusions acides (bisulfate, bifluorure) ou alcalines (carbonates, soude, peroxyde de sodium). L’attaque à haute température par le chlore gazeux en présence de carbone est également très efficace (la loparite est traitée de cette façon dans certains pays); ce procédé permet la séparation de certains lanthanides et d’autres éléments (fer, uranium, thorium) par volatilisation fractionnée des chlorures.Isolement du groupeAprès la décomposition des minerais, les lanthanides, mis en solution, sont séparés des éléments étrangers qui les accompagnent par diverses réactions de précipitation, exploitant l’insolubilité des hydroxydes, fluorures, oxalates, phosphates ou sulfates doubles alcalins. Cette dernière opération est souvent utilisée en outre pour réaliser un premier fractionnement grossier entre lanthanides légers (groupe cérique) et lanthanides lourds (associés à l’yttrium).Industriellement, alors que le traitement de la bastnaésite ne pose guère de problèmes (la concentration fournissant directement un mélange d’oxydes de lanthanides), celui de la monazite est plus complexe, car il implique la récupération de l’acide phosphorique et la séparation du thorium. Dans le procédé d’attaque sulfurique , le minerai traité par un excès d’acide concentré vers 200-250 0C donne une pâte de sulfates anhydres qui est reprise par l’eau, soit en quantité limitée pour n’en extraire que le sulfate de thorium, soit en grande quantité et à froid pour dissoudre la totalité des sulfates; dans cette seconde méthode, la plus courante, le thorium est séparé par précipitation sélective du phosphate, du fluorure, de l’oxalate ou de l’hydroxyde, en jouant sur l’acidité de la solution; on a recours également très souvent à la précipitation des lanthanides à l’état de sulfates doubles par addition de sulfate de sodium (dans ce cas, le thorium reste en solution avec les terres yttriques). Dans le procédé d’attaque alcaline , mis au point par l’industrie française, le minerai (monazite ou xénotime) est pulvérisé et chauffé vers 150 0C avec une lessive de soude concentrée, généralement en autoclave. Le produit de réaction est repris par l’eau et les hydroxydes de terres rares et de thorium sont séparés par décantation et filtration de la solution, dont on extrait le phosphate trisodique par concentration. On sépare le thorium des lanthanides, soit par dissolution sélective de ces derniers par l’acide chlorhydrique, soit par précipitation basique après dissolution nitrique de l’ensemble. Les deux techniques donnent directement un précipité d’hydroxyde de thorium (et d’uranium) contenant très peu de lanthanides, et une solution de lanthanides exempte de thorium, ce qui constitue l’avantage principal de l’attaque alcaline par rapport à l’attaque sulfurique.Quelle que soit la méthode utilisée pour la séparation du groupe des terres rares, on obtient finalement une solution (ou un précipité) contenant à la fois le mélange des lanthanides, leur homologue léger l’yttrium et parfois aussi le scandium; seules les techniques de séparation applicables aux lanthanides permettront, en cours d’opération, l’extraction de ces éléments.Séparation individuelleLa séparation des lanthanides les uns des autres reste encore à l’heure actuelle un problème difficile du point de vue économique, malgré l’efficacité des techniques d’échanges d’ions et d’extraction par solvants; cela tient à la répartition très inégale des différents éléments du groupe dans leurs mélanges naturels, et au fait que quelques-uns d’entre eux seulement trouvent des applications, tandis que les autres constituent une masse importante de sous-produits qui grève le prix des opérations.Certains lanthanides peuvent toutefois être séparés dans de bonnes conditions par des opérations chimiques dites «classiques». Il s’agit, d’une part, des éléments pouvant prendre des valences différentes de 3, d’autre part, de ceux qui sont particulièrement abondants dans les mélanges naturels. Les premiers peuvent être isolés par des méthodes d’oxydation ou de réduction sélective, les seconds extraits, du moins partiellement, par les méthodes de fractionnement (précipitation ou cristallisation).OxydoréductionL’oxydation sélective s’applique de façon parfaite à la séparation du cérium qui, à l’état tétravalent, est beaucoup moins basique que les autres lanthanides. Si l’on part d’une solution, on fait réagir un réactif oxydant tel que le persulfate, le bromate ou le permanganate, en milieu peu acide, puis on insolubilise le cérium IV par hydrolyse sous forme d’un sel basique, ou bien on extrait sélectivement par un solvant organique tel que le tributyl-phosphate. Mais, très souvent, on procède inversement à l’oxydation des hydroxydes par chauffage à l’air entre 100 et 150 0C, puis à la dissolution sélective des lanthanides trivalents par une solution d’acide nitrique diluée. Cette opération peut être réalisée en une fois en traitant une suspension des hydroxydes par le chlore gazeux. L’oxyde cérique hydraté, resté insoluble après ces traitements, peut être redissous dans l’acide nitrique concentré, et purifié par cristallisation sous forme de céri-nitrate d’ammonium. Ces procédés sont utilisés industriellement pour la production des sels de cérium, mais ils ne s’appliquent pas au praséodyme et au terbium qui ne peuvent être séparés sous forme de sels tétravalents que par oxydation électrolytique en sels fondus.La réduction sélective est une méthode particulièrement intéressante pour l’extraction de l’europium, élément peu abondant mais très recherché. La réduction s’effectue par action du zinc sur une solution chlorhydrique, et l’europium II, dont le comportement chimique est analogue à celui des alcalino-terreux, est précipité sous forme de sulfate en présence de baryum comme entraîneur; on le récupère en lavant le précipité par une solution oxydante acide, dans laquelle le sulfate de baryum reste évidemment insoluble. Les autres lanthanides réductibles à la valence + 2 (samarium et ytterbium) ne peuvent être isolés par cette méthode, qui est donc sélective pour obtenir l’europium. Par contre, l’utilisation d’amalgames de métaux alcalins permet la réduction des trois éléments jusqu’à l’état métallique, et leur extraction de l’ensemble des autres lanthanides; deux techniques sont utilisées, l’une par circulation à contre-courant d’amalgame de sodium et d’une solution chlorhydrique de lanthanides, l’autre par électrolyse sur cathode d’amalgame de lithium. Une autre méthode très efficace pour la séparation des éléments «bivalents» est la réduction des oxydes par le carbone, suivie d’une volatilisation sélective à haute température.Fractionnements successifsAprès l’extraction du cérium et éventuellement des éléments réductibles (Sm, Eu et Yb), les mélanges de terres rares ne contiennent plus que des éléments de propriétés chimiques très voisines, qui ne peuvent être séparés les uns des autres que par des méthodes de fractionnements successifs. En considérant deux éléments voisins, l’efficacité d’un fractionnement s’évalue par le rapport de leurs concentrations relatives dans le mélange avant et après l’opération. Ce rapport, appelé «facteur de séparation», dépend évidemment de la variation dans la série des lanthanides, de la propriété utilisée pour l’opération.La méthode des cristallisations fractionnées fut certainement la plus utilisée autrefois pour la séparation des terres rares; elle est fondée sur les différences de solubilité dans l’eau des sels de lanthanides tels que les nitrates doubles magnésiens, ammoniacaux ou manganeux, les bromates, les sulfates et les éthylsulfates. En concentrant une solution de sels isomorphes jusqu’à cristallisation partielle, les cristaux sont enrichis en éléments dont les sels sont moins solubles, et la solution en corps plus solubles; on peut répéter l’opération sur les deux fractions obtenues, et poursuivre les fractionnements jusqu’à l’obtention de produits purs. Cela nécessite malheureusement un très grand nombre d’opérations, et les quantités mises en jeu deviennent de plus en plus petites, tandis que les fractions se multiplient. Dans la pratique, on regroupe les fractions de compositions voisines, et on se limite à un nombre modeste d’opérations pour préparer des fractions enrichies en certains éléments abondants, par exemple des concentrés de néodyme, praséodyme ou lanthane dans le groupe des terres cériques.Les précipitations fractionnées exploitent les différences d’insolubilité observées pour certains composés de lanthanides en présence de quantités limitées de réactif précipitant. Ainsi, les hydroxydes précipitent dans l’ordre de leur basicité croissante, et les premiers précipités seront donc enrichis en lanthanides lourds; pour obtenir une précipitation sélective, il faut travailler en solution diluée, par exemple en faisant barboter de l’ammoniac gazeux mélangé à de l’air dans une solution de nitrates; on peut de cette façon obtenir des précipités de lanthane pratiquement pur. La précipitation des sulfates doubles avec le sodium, obtenue par addition progressive de sulfate de sodium à une solution de sulfate de lanthanides, fournit inversement des précipités d’abord riches en terres cériques, puis en terres yttriques; c’est l’une des méthodes les plus employées industriellement pour la séparation des deux sous-groupes de lanthanides. La précipitation fractionnée des oxalates en milieu complexant (acide éthylène-diamine-tétra-acétique, ou acide nitrilotriacétique) suit le même ordre et permet des séparations efficaces, par exemple celle des terres rares et du scandium (celui-ci précipitant en dernier).Échanges d’ionsLes techniques d’échanges d’ions sont à l’origine du développement spectaculaire de l’utilisation des lanthanides dans la recherche et l’industrie. La possibilité de séparer les cations des terres rares par adsorption (fixation) puis désorption (élution) sur échangeurs d’ions était connue depuis longtemps, mais le procédé n’a pris de l’importance qu’après la Seconde Guerre mondiale, quand les chercheurs américains travaillant au problème de la séparation des lanthanides, produits de fission de l’uranium, eurent l’idée d’utiliser comme échangeurs d’ions des «résines» synthétiques (polymères sulfonés) à grande capacité d’absorption, et comme éluants des solutions d’agents complexants (citrate d’ammonium en solution diluée à pH contrôlé). Le facteur essentiel de l’efficacité de la séparation est la différence de stabilité des complexes dans la série des lanthanides, stabilité qui varie en raison inverse de la basicité; en mettant une solution complexante au contact d’un échangeur d’ions sur lequel les cations de terres rares ont été préalablement fixés, on provoque une désorption préférentielle des éléments dont les complexes sont les plus stables, et la solution s’enrichit en ces éléments. En pratique, pour donner à l’opération son efficacité maximale, on opère sur des «colonnes», tubes verticaux remplis de granulé de résine échangeuse. Le mélange à séparer ayant été fixé à la partie supérieure de la colonne (entrée) par injection d’une solution de chlorures ou nitrates, le passage de la solution éluante provoque une cascade de désorptions et de ré-adsorptions des ions lanthanides, équivalentes à la succession d’un nombre considérable de fractionnements. Cette élution a pour effet d’entraîner les lanthanides à travers la colonne avec des vitesses de migration différentes, ce qui aboutit à la séparation des constituants du mélange, qui apparaissent finalement dans l’«éluat» dans l’ordre inverse de leurs basicités, c’est-à-dire de leurs numéros atomiques. En fractionnant les solutions à la sortie de la colonne à l’aide d’un dispositif collecteur (associé éventuellement à un appareil de contrôle), on matérialise la séparation, qui est évidemment plus ou moins complète suivant les conditions opératoires. En travaillant sur de très petites quantités de mélanges, avec des pH d’élution correspondant au seuil de formation des complexes (et éventuellement un gradient de pH), il est possible de réaliser des séparations totales par élution sélective des éléments les uns après les autres; ces conditions sont appliquées dans des buts analytiques, en utilisant principalement comme agents complexants l’acide citrique, l’acide lactique ou l’acide hydroxy-isobutyrique. Mais elles sont inapplicables pour la production des lanthanides en quantités importantes. Ce n’est que grâce à des améliorations à la fois dans les techniques opératoires et dans le choix des complexants que cette production a pu atteindre le stade industriel. L’inconvénient majeur de la technique d’élution sélective étant la dilution des solutions (ce qui nécessiterait dans l’industrie la manipulation de volumes prohibitifs de liquides), on lui a substitué des techniques de déplacement et de localisation. Dans le procédé mis au point au Laboratoire des terres rares du C.N.R.S., on opère sur une batterie de plusieurs colonnes. La solution éluante parcourant ces colonnes reliées en série y répartit les lanthanides dans l’ordre de leurs vitesses de déplacement, puis les différents éléments localisés dans chaque colonne sont purifiés et extraits par circulation «en parallèle» d’une solution éluante plus concentrée. D’autre part, des agents complexants plus efficaces que l’acide citrique ont permis d’améliorer la qualité des séparations: le plus utilisé est l’acide éthylène-diamine-tétra-acétique (EDTA), bien que sa faible solubilité oblige souvent à fixer au préalable sur les résines des ions échangeurs autres que l’hydrogène, par exemple Cu2+ ou Zn2+. On emploie aussi l’acide nitrilo-triacétique, l’acide hydrazine-diacétique (HDA) et les acides poly-amino-polycarboxyliques analogues à l’EDTA, tels que l’acide hydroxy-éthyl-éthylène-diamine-triacétique (HEDTA), l’acide di-éthylène-triamine-penta-acétique (DTPA), l’acide 1,2-diamine-cyclo-hexane-tétra-acétique (DCTA ou OCTA). Le choix judicieux de ces complexants en fonction de la composition des mélanges à traiter permet de résoudre pratiquement les problèmes les plus difficiles, y compris celui de la séparation de l’yttrium qui accompagne les lanthanides, mais qui peut-être localisé soit normalement avec les terres yttriques (acides acétique, EDTA), soit avec les terres cériques (acides HDA, HEDTA, DTPA).Les extractions par solvantsDe même que dans le cas des échanges d’ions, la possibilité de séparer les lanthanides par extraction sélective à l’aide de solvants organiques était connue bien longtemps avant que le choix de réactifs et de conditions convenables et la mise en œuvre de techniques continues n’en fassent une méthode de production vraiment efficace. Si une solution aqueuse de sels de lanthanides est mise en contact avec un solvant organique approprié, la substance se répartit entre les deux phases liquides, et le coefficient de partage (rapport des concentrations dans la phase organique et dans la phase aqueuse) varie avec le numéro atomique du lanthanide; la phase organique s’enrichit, en général, en éléments lourds. Les facteurs de séparation (rapport des coefficients de partage d’un élément au suivant) ne sont toutefois pas très élevés, de sorte que la séparation des constituants d’un mélange nécessite de nombreuses extractions successives, que l’on réalise en faisant circuler la solution aqueuse de sels et le solvant organique à contre-courant, dans une batterie d’extracteurs.Les solvants organiques initialement utilisés étaient des éthers, des cétones et 廓-dicétones, des alcools tels que le butanol; mais c’est avec le n -tributylphosphate (n -C4H9O)3P4 (TBP) que les premiers résultats marquants furent obtenus (D. F. Peppard, 1953), et c’est encore ce solvant qui est le plus utilisé (fig. 1), à côté d’autres alkyl-phosphates tels que l’acide di-(2-éthylhexyl)-ortho-phosphorique (acide HDEHP) qui sert à extraire des concentrés d’europium à partir des mélanges de lanthanides de la bastnaésite. L’acide HDEHP, très visqueux, doit toutefois être dilué dans un solvant non actif (toluène, éther de pétrole, etc.) tandis que le TBP peut être utilisé pur. Une autre classe importante de solvants est celle des amines et de leurs sels, en particulier les amines primaires à longues chaînes, que l’on emploie diluées dans du kérosène; l’extraction par ces solvants peut être considérée comme une sorte d’échange anionique, et les facteurs de séparation se trouvent sensiblement améliorés par addition, à la phase aqueuse, de complexants tels que les acides amino-polyacétiques (EDTA, DCTA, DTPA). Parmi les extractants des lanthanides d’utilisation récente, il faut citer enfin les acides naphténiques de formule générale R-COOH (où R est un radical cyclique de poids moléculaire élevé) qui s’emploient également avec des diluants et en association avec des complexants.Les sels de terres rares utilisés sont presque toujours des nitrates, bien que certains sels complexes tels que les citrates aient donné des résultats encourageants. Les nitrates de lanthanides sont souvent additionnés de nitrates alcalins, pour obtenir un effet de relargage. En tout cas, le facteur de séparation n’est élevé qu’avec des solutions aqueuses de force ionique élevée, c’est-à-dire fortement acides et très concentrées. Cette dernière condition est évidemment favorable au traitement de grandes quantités de produits et explique le développement qu’a pris rapidement le procédé d’extraction par solvants pour la séparation des lanthanides à l’échelle industrielle.Les procédés modernes de traitement des minerais et de séparation des lanthanides (fig. 2) ont atteint une grande efficacité, grâce à la combinaison des différentes techniques qui viennent d’être décrites.4. Propriétés physiquesStructure électroniqueLes lanthanides correspondent à l’irruption, dans la classification périodique des éléments, des électrons f , c’est-à-dire ceux pour lesquels le nombre quantique orbital l est égal à 3. Naturellement, cela n’est possible que lorsque le nombre quantique principal n atteint 4 (puisque l 諒 n 漣 1). La fonction d’onde d’un électron est le produit d’une partie radiale (qui dépend des nombres quantiques n et l ) et d’une partie orbite (qui dépend de l et des deux autres nombres quantiques m 1 (face=F0019 漣 l 諒 m 1 諒 + l ) et m s, nombre quantique de spin qui vaut + 1/2 ou 漣 1/2). Dans le cas de la fonction radiale 4f , il se trouve que le maximum de la courbe qui définit la probabilité de présence de l’électron en fonction de la distance au noyau, se trouve situé plus près du noyau que pour les couches 5s et 5p , complètement remplies pour le xénon, avant que les électrons f ne se manifestent. Au niveau du lanthane, la configuration électronique d’énergie la plus basse de l’atome neutre est [Xe]6s 25d 1. Comme les électrons périphériques sont facilement perdus lorsqu’une liaison chimique se crée, ou lorsque les atomes neutres se condensent en métal, le lanthane forme donc des ions trivalents, comme le scandium et l’yttrium qui se trouvent dans la même colonne du tableau de Mendeleïev et qui forment avec le lanthane et les lanthanides le groupe des terres rares .C’est au niveau de l’élément suivant, le cérium, que l’électron f apparaît dans la configuration de base de l’atome neutre qui est [Xe]6s 25d 14f 1. Le cérium peut former des ions trivalents par la perte des deux électrons 6s et de l’électron 5d , mais aussi des ions quadrivalents par la perte de l’ensemble des électrons supplémentaires par rapport au «cœur» xénon. Il en résulte que le métal cérium possède des propriétés très curieuses, car les atomes métalliques peuvent changer de valence et passer par exemple de + 3 à + 4 par cession partielle de l’unique électron f à la bande de conduction si le métal est refroidi. Comme les rayons d’ions sont différents, cette transformation se marque par un grand changement de volume, une contraction avec effet d’hystéresis, et une modification des propriétés physiques à basse température entre 200 et 100 K.Au niveau du praséodyme, la configuration de base de l’atome neutre (déterminée expérimentalement) est [Xe]6s 24f 3. On pourrait donc s’attendre à ce que le praséodyme forme des ions divalents par la perte des deux électrons 6s extérieurs à la structure xénon. En fait il n’en est rien; le praséodyme forme des ions trivalents et aussi dans certains cas des ions quadrivalents comme ceux du cérium. De même la configuration de base des atomes neutres de néodyme est [Xe]6s 24f 4, mais les ions sont toujours trivalents. Il se produit en effet un phénomène curieux. Les configurations excitées [Xe]6s 25d 14f 2, pour le praséodyme, et [Xe]6s 25d 14f 3 pour le néodyme, sont situées juste au-dessus de la configuration de base (fig. 3). Or, les positions relatives des configurations des atomes neutres sont modifiées lorsque ces atomes se condensent. Lors du passage de la vapeur métallique dans laquelle les atomes neutres sont dispersés au métal liquide ou solide, les configurations [Xe]6s 25d 14f N-1 deviennent configurations de base et, en conséquence, les ions sont trivalents par perte de l’électron 5d et des deux électrons 6s . Ce phénomène a lieu pour presque tous les lanthanides, à l’exception de l’europium et de l’ytterbium pour lesquels les configurations excitées contenant l’électron 5d sont situées assez haut en énergie (fig. 3). Dans ces deux cas, les atomes sont divalents dans la phase métallique et dans quelques composés. Cependant dans la plupart des combinaisons chimiques, pour ces éléments aussi, la valence trois prédomine. Dans le cas du gadolinium (milieu de la série) la configuration de base est [Xe]6s 25d 14f 7, et, par conséquent, le métal est «naturellement» trivalent (comme le lanthane et le lutétium).Expérimentalement, ce phénomène particulier se remarque au fait que le troisième potentiel d’ionisation des lanthanides (correspondant à l’énergie nécessaire au passage 4f N4f N-1 ) est quantitativement équivalent à la chaleur de sublimation du métal, c’est-à-dire à l’énergie nécessaire pour disperser les atomes neutres. Cette énergie varie beaucoup, en dents de scie, selon la valeur de N (nombre d’électrons 4f ). Les irrégularités de cette fonction sont la source des «accidents» (effet «tétrade») que les chimistes spécialisés trouvent quelquefois dans la variation des propriétés chimiques de certains composés trivalents des lanthanides. Ces propriétés sont généralement très voisines, elles varient d’ordinaire régulièrement, le seul paramètre qui induise les différences étant la diminution du rayon d’ion à mesure que le nombre d’électrons f croît (contraction lanthanidique), car le maximum de la fonction radiale f se rapproche du noyau à mesure que la couche se remplit. Les ions trivalents des lanthanides correspondent donc à des configurations de base de type [Xe]4f N , de même d’ailleurs que pour les ions divalents (Eu2+ et Gd3+ sont isoélectroniques: [Xe]4f 7). Cependant quelques-uns de ces ions montrent une particularité remarquable: la présence d’une configuration excitée située au-dessus de la configuration de base, accessible par des excitations dans la gamme de l’énergie des rayonnements électromagnétiques du domaine visible ou de l’ultraviolet proche. C’est le cas pour l’ion trivalent du cérium [Xe]4f 1, qui possède une configuration excitée [Xe]5d 1. La position de cette configuration dépend beaucoup de la nature du composé chimique dans lequel l’ion est engagé. Il en est de même pour la majorité des ions divalents qui peuvent être obtenus à l’état solide (configuration de base [Xe]4f N , excitée [Xe]4f N-1 5d 1). Comme les transitions optiques entre configurations de parités opposées sont permises, donc intenses, cette propriété joue un grand rôle dans les applications pratiques des lanthanides (luminophores à base de Ce3+ et Eu2+).Un grand domaine de recherche s’est développé autour de ce que l’on appelle les composés à valence variable des lanthanides. C’est une propriété qui découle de ce qui a été exposé ci-dessus. Pour certaines combinaisons chimiques, il se trouve que la configuration excitée 4f N-1 5d 1 est abaissée à la formation du solide, mais pas assez pour qu’une différence nette s’établisse avec la configuration de base de l’atome neutre, 4f N . L’atome va se trouver alors dans une situation où il possède, en quelque sorte, simultanément les deux valences. La physique du système devient très complexe et difficile à interpréter théoriquement. C’est le cas par exemple pour SmS et TmSe, ainsi que pour le cérium métal (cf. supra ).Propriétés spectralesSi la chimie des lanthanides est relativement insensible à la présence des électrons f , il n’en est pas de même de la physique des composés où sont engagés les atomes Ln3+ (Ln est un symbole général pour le groupe des lanthanides). En effet, les propriétés spectroscopiques et les propriétés magnétiques dépendent étroitement des différents états que peuvent prendre les électrons dans les configurations 4f N (N : nombre d’électrons 4f ).Les spectres obtenus à partir des atomes de terres rares, notamment en émission (plasmas d’arc et d’étincelle), sont particulièrement compliqués et leur interprétation théorique est l’un des plus beaux succès de la théorie quantique. L’ensemble constituant un atome, noyau et électrons, possède seulement un nombre limité et défini d’états énergétiques. Pour les atomes libres (non engagés dans une liaison chimique) et pour une configuration déterminée (n et l fixés), ces états sont des niveaux J; ils sont notés (2S+1)Lj [cf. SPECTROSCOPIE].L’effet du couplage spin-orbite qui définit les niveaux J est particulièrement important pour les atomes des terres rares. Le couplage réel est intermédiaire entre le couplage LS (Russell-Saunders) et le couplage j-j . Les expérimentateurs s’en tiennent cependant, pour l’identification des niveaux, au couplage LS.De nombreux niveaux, d’énergies supérieures, se succèdent à intervalles rapprochés entre le proche infrarouge et l’ultraviolet, notamment pour Nd3+, Sm3+, Dy3+ et Ho3+. Certains d’entre eux sont fluorescents, et quelques transitions ont une grande importance pratique, par exemple 5D072 de Eu3+ (fluorescence rouge de la télévision en couleurs) et 43/24I11/2 de Nd3+, émission à 1,06 猪m des lasers au néodyme.Lorsque l’on introduit un atome de terre rare dans un solide, la dégénérescence des niveaux J est levée grâce au champ cristallin et les niveaux se séparent (effet Stark) en 2J + 1 composantes au maximum si le nombre d’électrons f est pair ou en J + 1/2 composantes au maximum si le nombre d’électrons est impair. Cet effet du champ cristallin n’est pas très important. C’est une grande différence par rapport aux éléments à électrons d ; en effet, pour ceux-ci, l’effet du champ cristallin est beaucoup plus important que l’effet du couplage spin-orbite. Il en résulte que les atomes de terres rares conservent dans les solides des niveaux d’énergie bien définis. Cette propriété est très importante, car elle est à la base de toutes les applications pratiques en optique: les transitions entre les niveaux ont des longueurs d’onde bien définies. L’intensité des transitions dépend toutefois très fortement de la liaison chimique supportée par l’atome de terre rare et donc de la structure et de la nature du matériau.La décomposition des niveaux(2S+1)LJ par le champ cristallin dépend de la symétrie de celui-ci, donc de la symétrie du site ponctuel cristallographique occupé par la terre rare. Le nombre de transitions optiques observables dépend des valeurs de J des niveaux inférieurs et supérieurs. Il est fixé par les règles de la théorie des groupes. Cette propriété permet d’utiliser les lanthanides comme sondes structurales ponctuelles dans les matériaux minéraux, organiques, biologiques et même en solution liquide. Certains lanthanides ont des niveaux d’énergie, ou des transitions, bien adaptés à ce rôle. Ce sont principalement le néodyme, l’europium, le gadolinium et le terbium. L’apparition des lasers à colorants qui permettent d’exciter directement un niveau d’énergie précis a grandement développé l’emploi des lanthanides pour la compréhension des problèmes d’ordre locaux dans les structures cristallines complexes et les milieux biologiques (fig. 4). On peut ainsi déterminer par exemple si le lanthanide occupe plusieurs sites cristallographiques dans une phase donnée, ou faire des études de systèmes, le spectre optique étant alors l’analogue pour un site particulier d’un diagramme de poudre de rayons X pour un composé défini.Les configurations 4f N sont considérablement dégénérées. Le nombre d’états |SLJMJ 羚 différents est 14!/N !(14-N )! En situation de basse symétrie la dégénérescence peut être totalement levée (mais si n est impair, les niveaux sont des doublets de Kramers). L’interprétation quantitative des spectres optiques se fait en développant le hamiltonien du champ cristallin sous la forme d’une somme d’opérateurs tensoriels monoélectroniques. Le rang des tenseurs est pair et inférieur ou égal à 6. Les éléments de matrice se calculent avec les techniques de l’algèbre de Racah. Les coefficients des opérateurs, appelés paramètres du champ cristallin, peuvent être déterminés en appliquant l’hamiltonien sur la base |SLJMJ 羚 la plus étendue possible (ce qui fait par exemple pour 4f 3 une matrice 364 憐 364) et en comparant le résultat du calcul à l’expérience. Un grand nombre de ces déterminations empiriques ont été faites. Les paramètres changent peu pour une même structure, d’une terre rare à l’autre. Cependant, les théoriciens tentent de calculer a priori les paramètres du champ cristallin à partir de la connaissance de la structure en utilisant d’une part une approche électrostatique (charges ponctuelles et multipôles induits à travers le cristal) et d’autre part, une description du recouvrement orbital au niveau de l’environnement local du lanthanide. Les résultats sont assez satisfaisants et ouvrent la voie à une prédiction des propriétés optiques des lanthanides dans les solides (et aussi des propriétés paramagnétiques), ce qui est important pour certaines applications (lasers de puissance). C’est aussi un exemple concret de l’orientation de la science moderne vers le triptyque: expérience-théorie-simulation, qui conduit à réaliser de véritables expériences sur ordinateur (en explorant par exemple les propriétés de structures virtuelles non encore réalisées par le chimiste).Propriétés magnétiquesLes niveaux (2S+1)Lj de base des atomes de terres rares Ln3+ indiqués au tableau 2 suivent la règle de Hund. Les systèmes à N électrons f (N : 0,..., 7) du lanthane au gadolinium sont analogues aux systèmes à 14 漣 N «trous» dans la couche 4f (terbium au lutétium), et les atomes possèdent les mêmes niveaux J, mais dans un ordre différent pour chaque terme: le niveau fondamental correspond à J = L 漣 S de La à Eu, à J = L + S de Tb à Lu.En présence d’un champ magnétique, la dégénérescence des niveaux J est levée et ils se séparent en 2 J + 1 composantes. En principe, à température ordinaire, seul le niveau fondamental est peuplé. Le moment magnétique de l’atome est donné par 猪 = gJ 猪B avec 猪B = magnéton de Bohr, g = facteur de Landé, soit:
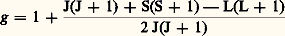 Du fait de la règle de Hund, les propriétés magnétiques ne sont pas symétriques dans la série de part et d’autre du gadolinium, mais tous les atomes possèdent un moment magnétique donné par la formule ci-dessus et en bon accord avec l’expérience, à l’exception toutefois du samarium et de l’europium pour lesquels les composantes du multiplet (niveaux J du terme) ne sont pas suffisamment séparées et présentent, avec le niveau de base des différences de l’ordre de kT (kT à température ordinaire vaut 210 cm-1). Ces atomes possèdent un moment magnétique qui se calcule différemment (formule de Van Vleck).Il en résulte que, à l’exception du lanthane et du lutétium, tous les composés des terres rares sont paramagnétiques, et en général (sauf les composés de Eu3+ et Sm3+) suivent la loi linéaire de Curie-Weiss pour la variation du paramagnétisme avec la température.L’interaction entre atomes magnétogènes se traduira pour certains composés par l’apparition d’un ordre magnétique : ferromagnétisme, ferrimagnétisme, antiferromagnétisme, hélimagnétisme. Cette possibilité est très utilisée pratiquement. Par exemple, l’oxyde EuO (oxyde de Eu2+ équivalant électroniquement à Gd3+) est ferromagnétique (moments magnétiques alignés parallèlement), de même certains alliages comme Gd3Al2, Dy3Al2, etc., et surtout les alliages terres rares-cobalt du type Ln2Co17 et LnCo5 qui ont en outre des moments de saturation importants et des points de Curie très hauts (1 000 0C). Les substances ferrimagnétiques sont constituées de deux ou plusieurs sous-réseaux alignés de façon anti-parallèle avec des aimantations inégales en valeur absolue (théorie de Néel). C’est le cas des ferrites de lanthanides LnFe3 (pérovskites) et Ln3Fe512 (grenats) qui sont utilisées pour fabriquer des semiconducteurs.5. Propriétés chimiquesChimie en solutionLa chimie des lanthanides en solution est caractérisée par la prédominance quasi exclusive de la valence + 3 (la seule exception est l’europium, Eu2+), et par des réactions chimiques analogues tout au long de la série. Les ions en solution sont coordonnés par neuf molécules d’eau formant le complexe [Ln(H2O)9]+3. Certains sels (chlorures, nitrates) sont très solubles, d’autres très insolubles, notamment les oxalates (ceux-là, même en milieu acide: réaction de caractérisation et séparation des lanthanides en chimie analytique), les carbonates, les hydroxydes, les phosphates, les fluorures. La solubilité des composés peu solubles varie dans la série d’une façon irrégulière, par exemple la solubilité des hydroxydes décroît du lanthane au lutétium, elle croît pour d’autres sels (carbonates, sulfates), et, pour certains, elle passe par un maximum au niveau du gadolinium (glycollates). Les lanthanides en solution donnent de nombreux complexes avec les diacides organiques, et cela est utilisé pour les séparations. Les potentiels d’électrode E0(Ln3+aq /Ln) sont très négatifs, variant de – 2,52 V pour le lanthane à – 2,25 V pour le lutétium. Eu2+ en solution est analogue aux éléments alcalino-terreux et présente des réactions très voisines de celles de Sr2+.Chimie à l’état solideLa chimie de l’état solide des lanthanides est beaucoup moins monotone que la chimie en solution. Les structures cristallines sont souvent, mais pas toujours, identiques pour un composé donné, tout au long de la série. En général, dans ce cas, les paramètres du réseau décroissent de La à Lu, en raison de la contraction lanthanidique . En effet, à mesure que l’on ajoute des électrons f à l’atome, le rayon ionique de Ln3+ diminue. Il doit d’ailleurs être défini par rapport à un atome donné, car il est relativement variable: on obtient des valeurs différentes, par exemple, suivant que l’on part de la série des oxydes ou de celle des chlorures. Pour les oxydes, en prenant comme rayon ionique pour l’oxygène r = 0,138 nm, on obtient, d’après les distances Ln-O, des rayons qui varient de 0,102 nm pour le lanthane à 0,083 nm pour le lutétium.C’est dans l’état solide que l’on observe le plus facilement les autres valences des lanthanides. La valence +4 est obtenue pour le cérium, le praséodyme et le terbium dans les oxydes notamment, quelquefois pour d’autres éléments dans les fluorures complexes. La valence +2 est observée d’une façon très générale dans les systèmes métaux-halogénures et pour l’europium dans les oxydes. Les valences +2 sont également obtenues pour Eu dans certains sels (sulfates, carbonates).Les oxydes sont des composés très réfractaires qui fondent vers 2 200 0C. Ils possèdent trois structures cristallines principales, A hexagonale, B monoclinique et C cubique. Les structures A et B sont des phases de haute température, les températures de transformation montent dans la série. Les structures de ces oxydes, surtout A et B, n’ont pas d’équivalent pour d’autres éléments. En effet, elles reposent sur une coordination tétraédrique de l’oxygène par des atomes de terres rares. Les tétraèdres OLn4 ainsi définis sont joints par leurs arêtes et forment dans les structures A et B un véritable ion complexe à symétrie ternaire (LnO)nn +. Ces oxydes doivent donc se formuler (LnO)2O. La forme C-Ln23 est un assemblage tridimensionnel de tétraèdres OLn4 joints par quatre de leurs arêtes sur six. Le cation (LnO)nn + se rencontre aussi dans de très nombreux oxysels: oxychlorures (LnO)Cl, oxycarbonates (LnO)2C3, oxysulfures (LnO)2S, etc. L’arrangement des tétraèdres peut être aussi de symétrie quaternaire (LnOCl). Ces oxysels sont complètement insolubles, assez inertes chimiquement et généralement réfractaires, ils sont à la base de la variété observée dans la chimie de l’état solide par rapport à la chimie en solution. Ils forment aussi les meilleures matrices pour l’exploitation des propriétés optiques des ions Ln3+, du fait de l’influence de la liaison partiellement covalente Ln-O sur l’intensité des transitions.Les oxydes non stœchiométriques Cex, Prx et Tbx dus au mélange des valences +4 et +3 sont des exemples classiques de composés non stœchiométriques. Ils présentent de nombreuses phases intermédiaires, notamment pour des valeurs de x égales à 1,714, 1,778, 1,800, 1,833.Les atomes de terres rares entrent aussi dans la composition de nombreux oxydes mixtes avec d’autres éléments. Là encore, ils ont souvent tendance à coordonner tétraédriquement les atomes d’oxygène, par exemple dans les pyrochlores.Les chlorures, bromures et iodures anhydres sont extrêmement sensibles à l’humidité et ne peuvent être manipulés qu’en boîte à gants. On rencontre deux formes cristallines dans la série, une forme 見 type UCl3 (coordination 9 de la terre rare), et une forme 廓 type AlCl3 (coordination 6); la forme 見 au début, la forme 廓 à la fin de la série. Certains de ces halogénures ont aussi une structure du type PuBr3. Les points de fusion (600 à 900 0C) ont un minimum vers le milieu de la série. Les fluorures, beaucoup moins sensibles à l’humidité, sont plus réfractaires (point de fusion 1 500 à 1 200 0C, décroissant dans la série) et ont aussi deux structures La3 (hexagonale) au début de la série, Y3 (orthorhombique) après le gadolinium. Les chlorures fondus au contact du métal permettent d’obtenir pour presque tous les éléments les chlorures divalents très sensibles à l’oxydation.Les hydrures, borures, carbures, nitrures et sulfures sont des éléments d’un caractère beaucoup moins «ionique». Les hydrures LnHx sont non stœchiométriques, les borures ont pour formules LnB4 et LnB6, les carbures Ln3C, Ln2C3, les nitrures LnN, les sulfures Ln2S3 et LnS2. Ces substances présentent probablement des liaisons métaux-métaux, elles sont généralement d’une couleur très sombre et sont quelquefois des semiconducteurs ou même des conducteurs métalliques. Les lanthanides, dans ces composés, ont un comportement assez voisin de celui des éléments à électrons d , peut-être à cause de la participation d’électrons 5d aux liaisons.Les composés d’anions oxygénés sont très nombreux. Les hydroxydes ont une structure hexagonale où la terre rare est coordonnée par neuf ions OH-, les carbonates Ln2(CO3)3.n H2O (n = 8 au début de la série, 2 à 3 ensuite) et les nitrates Ln2(NO3)36H2O ont des structures caractérisées par une haute coordination de l’atome de terre rare, 10, 11 voire 12. Il en est probablement de même dans les autres sels hydratés, sulfates Ln2(SO4)3.8H2O, chlorures LnCl3.n H2O, n = 6 ou 7, etc., et les nombreux sels mixtes hydratés comportant d’autres cations.Les terres rares forment des complexes organiques assez nombreux, les plus intéressants étant les chélates de 廓-dicétones.La chimie de l’état solide est donc caractérisée par une grande variété: composés contenant un groupe à liaison partiellement covalente (LnO)nn +, composés à liaison métal-métal, sels beaucoup plus ioniques à haute coordination et composés de valence inhabituelle. Ces différences se voient dans la position des raies du spectre d’absorption de Ln3+, les raies sont déplacées vers le rouge pour les composés les plus covalents (oxydes, oxysels): c’est l’effet néphélauxétique.À haute température, il existe en outre des molécules LnO qui sont stables jusqu’à 6 000 K. On rencontre ces molécules dans certaines étoiles.MétallurgieLes métaux des terres rares sont préparés industriellement par la réaction du calcium avec le fluorure ou le chlorure anhydre à haute température et sous atmosphère inerte dans un creuset de tantale ou de tungstène. Les métaux ainsi obtenus sont en général assez purs au point de vue des éléments métalliques (Ca, Fe, Al, etc.) mais peuvent contenir des quantités notables d’impuretés non métalliques: hydrogène, oxygène, azote et carbone. Les métaux en couches minces sont de puissants getters , notamment pour l’hydrogène. Pour des expériences de recherche, il est nécessaire de purifier ces métaux, selon le cas, par refusion sous vide, sublimation, fusion de zone, ou électro-transport des impuretés à l’état solide.Les structures cristallines que présentent les métaux des terres rares sont très nombreuses. Leur description peut se faire au moyen des notations utilisées pour les matériaux polytypiques. Le tableau 3 donne un relevé des structures en fonction de la température, ainsi que le point de fusion et la pression de vapeur au point de fusion.Les électrons 4f demeurent localisés dans les lanthanides métalliques, les propriétés magnétiques sont donc celles des ions trivalents à l’exception de Eu et Yb, qui correspondent à des ions divalents, et du cérium qui est un cas de valence variable (entre 3 et 4). Ce sont surtout les lanthanides du groupe yttrique les plus lourds qui donnent lieu à des phénomènes d’ordre magnétique. Gd est ferromagnétique en dessous de 293,2 K 練 Tb, Dy, Ho et Tm ont deux structures magnétiques différentes dont l’une est simplement ferromagnétique et l’autre beaucoup plus complexe, les spins formant des hélices. Er possède trois structures magnétiques. Les complications sont dues au fait que l’axe de facile aimantation varie dans le cristal en fonction de la température.Ce, Y, Sc, Lu et surtout La sont supraconducteurs à haute pression. Pour La, la température critique est de 4,87 K à pression ordinaire et croît rapidement avec la pression (13 K à 200 kbar). Cependant, l’une des découvertes les plus remarquables est la mise en évidence de composés pour lesquels on observe simultanément un ordre magnétique et le phénomène de supraconductivité. C’est en particulier le cas pour les phases de Chevrel , GdMo6S8, TbMo6S8, qui sont à la fois supraconductrices et antiferromagnétiques à une température en moyenne inférieure à 2 K. Le composé ErRh4B4 présente une transition curieuse d’un état supraconducteur à un état ferromagnétique à 0,9 K.Les alliages formés par les métaux de terres rares sont très nombreux. On retiendra surtout les alliages avec le cobalt du type LnCo5 et les alliages ternaires avec le fer et le bore qui ont d’importantes applications comme aimants permanents. Les alliages amorphes formés avec les éléments du groupe 3d présentent quelquefois, malgré l’absence de structure cristallographique, de curieux phénomènes d’ordre magnétique: les moments pointent dans des directions déterminées par un cône ou une demi-sphère. On peut former dans ces matériaux (GdCo, GdFe, TbFe, etc.) des domaines magnétiques, par exemple sous forme de bulles, ils pourraient par conséquent servir de support à des informations lues ou écrites par des techniques laser thermomagnétiques.6. UtilisationsL’industrie des terres rares date de la fin du XIXe siècle, et elle a d’abord consommé essentiellement, soit des mélanges provenant de la monazite après extraction du thorium, soit des composés d’éléments faciles à séparer comme le cérium et le lanthane. Ce n’est que beaucoup plus tard, vers le milieu du XXe siècle, que se sont développées les utilisations basées sur les propriétés spécifiques de certains lanthanides (propriétés électroniques et spectrales en particulier).ComposésLes industries du verre et de la céramique comptent parmi les principales consommatrices de lanthanides. Des tonnages très importants d’oxydes de terres cériques, et plus spécialement d’oxyde de cérium, sont vendus sous forme de poudres pour polissage du verre (glaces, miroirs, prismes, lentilles et verres correcteurs d’optique médicale). Mais les oxydes de lanthanides interviennent aussi directement dans la fabrication de certains verres spéciaux: verre au lanthane à haut indice de réflexion et faible dispersion, verres de contraste au didyme (mélange de néodyme et praséodyme), verres protecteurs au néodyme absorbant les radiations jaunes des lampes au sodium, verres filtrants au cérium absorbant l’ultraviolet (verres de Crooks); l’oxyde de cérium, par son pouvoir oxydant, est aussi un excellent décolorant et stabilisant, il permet de préparer des verres de grande clarté et résistant aux radiations énergétiques (rayons X, rayons 塚). On emploie également les oxydes de lanthanides pour la verrerie d’art, en exploitant la couleur propre de leurs ions, soit pour compenser des colorations parasites (l’oxyde de néodyme est beaucoup employé dans ce but), soit pour obtenir des verres colorés (violet, rose ou pourpre avec le néodyme, vert avec le praséodyme, doré avec le cérium associé au titane).L’oxyde de cérium a été également beaucoup utilisé comme opacifiant pour les émaux, mais il est concurrencé par l’oxyde de titane, moins cher. L’oxyde de praséodyme sert à préparer un émail à base de silicate de zirconium d’un jaune intense, très apprécié pour la coloration de la porcelaine, et d’autres pigments contenant des terres cériques ont été mis au point récemment. L’emploi des oxydes de lanthanides a pris surtout de l’importance dans le domaine des céramiques industrielles: émaux et revêtements réfractaires, bougies d’allumage, et surtout céramiques diélectriques contenant par exemple du titanate ou du stannate de lanthane, à faible coefficient de température, utilisées dans les circuits résonnants (tels les oscillateurs de missiles). Les oxydes de terres rares entrent aussi, avec ceux de zirconium et de thorium, dans la composition d’éléments chauffants, conducteurs à haute température et résistant aux oxydants.L’industrie des catalyseurs a permis également aux lanthanides un débouché considérable, avec la mise au point de catalyseurs à base de terres rares pour le cracking du pétrole; ces catalyseurs améliorent le rendement en fractions légères du cracking et sont peu coûteux, car on les fabrique à partir de zéolites (alumino-silicates) et de mélanges de terres cériques (La, Nd, Pr). Cette application a pris aux États-Unis un développement très rapide et consomme, en tonnage, plus de la moitié de la production américaine d’oxydes de lanthanides; en Europe, par contre, ces catalyseurs sont beaucoup moins employés. Des catalyseurs à base de terres rares ont été brevetés pour de nombreuses autres réactions: polymérisations, isomérisations, hydrogénations et déshydrogénations, etc. Signalons, en particulier, l’oxydation catalytique de l’acide chlorhydrique pour l’obtention du chlore (ancien procédé Deacon) et l’utilisation de l’oxyde de cérium dans les moteurs d’automobile pour améliorer la combustion de l’essence.Une utilisation d’avenir est celle du cobaltite de lanthane comme catalyseur de réduction de l’oxygène dans les piles à combustibles des véhicules électriques.Les industries de l’éclairage sont probablement le domaine où l’utilisation des terres rares est le plus solidement implantée et offre les meilleures perspectives de développement, grâce aux propriétés spectrales spécifiques des lanthanides. Les propriétés émissives de l’oxyde de cérium, additionné en petites quantités à l’oxyde de thorium, ont fait le succès des «manchons Auer» d’éclairage au gaz; les «bâtonnets Nernst» sont toujours utilisés, comme sources de lumière très stables riches en infrarouges. Les oxydes et fluorures de lanthanides entrent depuis longtemps dans la fabrication des charbons à arc électrique intensif pour projecteurs; cette application consomme actuellement de 10 à 15 p. 100 de la production mondiale de lanthanides, principalement pour l’équipement des studios et salles de projection de cinéma, les équipements militaires, etc. Mais une application nouvelle a pris en peu de temps une importance considérable: c’est l’utilisation des lanthanides, et tout particulièrement de l’europium, dans les «phosphors», c’est-à-dire dans les matériaux luminescents employés dans les lampes d’éclairage. Dans ces substances, le lanthanide émetteur est introduit en petites quantités dans une «matrice» convenable, et l’émission lumineuse est obtenue par excitation à l’aide de radiations ultraviolettes (photoluminescence). Suivant l’élément de lanthanide choisi comme activateur, l’émission lumineuse est localisée dans le proche ultraviolet (avec le gadolinium, le cérium), le visible (rouge avec l’europium, orange avec le samarium, vert avec le terbium, jaune avec le dysprosium, bleu avec le thulium), ou le proche infrarouge (avec le néodyme). Cette propriété est utilisée pour améliorer la répartition spectrale des tubes fluorescents, et surtout dans les lampes à vapeur de mercure dans lesquelles un «phosphor» à base de terres rares, le vanadate d’yttrium activé à l’europium, transforme l’émission ultraviolette du mercure en lumière visible; ces lampes, qui donnent une lumière comparable à la lumière du jour, sont de plus en plus utilisées dans l’éclairage public. L’utilisation du vanadate d’yttrium activé à l’europium en cathodoluminescence (excitation par les électrons), pour les écrans de télévision en couleurs, a littéralement bouleversé l’industrie des terres rares dans les années soixante et, bien que le vanadate ait été supplanté par d’autres matrices (oxysulfures ou oxydes d’yttrium et de gadolinium), l’intérêt pour les propriétés spectrales de l’europium n’a pas diminué. Ce sont ces mêmes propriétés émissives qui sont exploitées, avec le néodyme, pour produire la lumière cohérente des lasers (verres ou monocristaux de grenat d’yttrium), et, avec divers lanthanides du groupe yttrique: Ho, Er, Yb, pour la construction de dispositifs transformant par des mécanismes de transfert d’énergie, les rayons infrarouges en lumière visible (compteurs quantiques).MétauxLes utilisations des lanthanides sous forme métallique consomment de 8 à 10 p. 100 de leur production globale. Elles font surtout appel à des mélanges, tels que le mischmétal (mélange des métaux cériques) utilisé en alliage avec 30 p. 100 de fer pour la fabrication des pierres à briquets. Cette industrie, qui date du début du siècle, est un monopole de producteurs de terres rares, et la production mondiale dépasse maintenant cinq cents tonnes par an, soit près de cinq milliards de pierres; l’application des propriétés pyrophoriques des alliages de lanthanides pour d’autres usages (pyrotechnie, signalisation, balles traceuses) reste modeste.Par contre, le mischmétal est de plus en plus utilisé en sidérurgie, comme désoxydant et désulfurant, pour la fabrication de fer ductile et d’aciers inoxydables à grains fins, pouvant être travaillés à chaud, et en fonderie, comme stabilisateur du carbone dans l’élaboration des fontes à graphite sphéroïdal: on peut dire qu’en Europe, pratiquement tous les alliages globulisants de la fonte contiennent du cérium additionné souvent sous forme d’un ferro-alliage spécial.Les métallurgistes utilisent aussi les métaux des lanthanides en alliages avec des métaux non ferreux, pour obtenir des propriétés améliorées: l’addition de mischmétal aux alliages nickel-chrome augmente leur résistance à la corrosion à chaud, et elle intervient dans la fabrication de presque toutes les qualités de fils électriques pour éléments chauffants: le céralumin (alliage aluminium-cérium) était utilisé dans la construction de pièces de moteurs d’avions dès la Première Guerre mondiale, et l’avènement de l’aviation supersonique et des fusées a beaucoup accru l’intérêt porté aux alliages légers améliorés aux terres rares: on observe une tendance à l’utilisation de métaux séparés de préférence aux mélanges, par exemple les alliages de magnésium-néodyme sont réputés pour leur excellente tenue mécanique à chaud.D’autre part, les propriétés magnétiques des métaux des lanthanides, qui n’avaient pas trouvé d’applications industrielles notables, sont passées au premier plan récemment avec la découverte des alliages de cobalt-lanthanides, de formule LnCo5 ou Ln2Co17, qui sont des aimants permanents plus puissants que tous ceux connus jusqu’à présent. On a fabriqué par exemple par frittage des aimants de samarium-cobalt (SmCo5) ayant des caractéristiques au moins deux fois plus élevées que les aimants de cobalt-platine industriels, et beaucoup moins chers que ces derniers; les performances théoriques de certains alliages cobalt-lanthanides (en particulier PrCo5) sont encore supérieures aux valeurs actuellement atteintes. Par ailleurs, la mise au point d’alliages ternaires Ln-Co-Fe ou Ln-Co-Cu préparés par fusion augmente encore l’intérêt de ces nouveaux aimants, utilisables non seulement pour les tubes micro-ondes et les autres dispositifs électroniques, mais également pour la construction des moteurs électriques.L’utilisation des lanthanides dans les technologies de pointe, magnétisme, électronique, sciences nucléaires, offre des perspectives d’avenir très variées:– Parmi les applications en cours de développement, on peut citer les masers et lasers (monocristaux de grenat d’yttrium-aluminium dopés au néodyme) utilisés en optique, télémétrie et télécommunications, les grenats ferrimagnétiques d’yttrium et de gadolinium, employés dans les dispositifs électroniques utilisant les micro-ondes, les orthoferrites (LnFeO3), les monochalcogénures d’europium (EuO, EuS, EuSe, EuTe) dont les propriétés magnéto-optiques sont exploitables pour des fonctions de mémoire ou de transmission dans les ordinateurs, les borures (de lanthane, en particulier) qui servent à fabriquer des cathodes émissives pour canons à électrons et tubes électroniques divers.– L’industrie nucléaire, d’autre part, utilise les très fortes sections efficaces d’absorption des neutrons (du samarium, du gadolinium, du dysprosium ou de l’europium) pour la fabrication de barres de contrôle et de réglage dans les réacteurs nucléaires, ou dans les revêtements de protection contre les radiations. Enfin, les radio-éléments artificiels des lanthanides (produits de fission ou obtenus par irradiation) offrent un large choix d’utilisations futures, basées sur leurs radiations: sources portatives de rayons X ou rayons 塚 pour radiographie et radioscopie, usages thérapeutiques, composés radio-luminescents activés au prométhium, micro-générateurs d’électricité par conversion d’énergie radio-voltaïque, etc.Toutes ces applications nouvelles, faisant appel à la spécificité des propriétés des lanthanides, permettent de penser que ces éléments prendront dans l’avenir une importance industrielle croissante, et expliquent que les spécialistes les considèrent comme les «matériaux du XXIe siècle».
Du fait de la règle de Hund, les propriétés magnétiques ne sont pas symétriques dans la série de part et d’autre du gadolinium, mais tous les atomes possèdent un moment magnétique donné par la formule ci-dessus et en bon accord avec l’expérience, à l’exception toutefois du samarium et de l’europium pour lesquels les composantes du multiplet (niveaux J du terme) ne sont pas suffisamment séparées et présentent, avec le niveau de base des différences de l’ordre de kT (kT à température ordinaire vaut 210 cm-1). Ces atomes possèdent un moment magnétique qui se calcule différemment (formule de Van Vleck).Il en résulte que, à l’exception du lanthane et du lutétium, tous les composés des terres rares sont paramagnétiques, et en général (sauf les composés de Eu3+ et Sm3+) suivent la loi linéaire de Curie-Weiss pour la variation du paramagnétisme avec la température.L’interaction entre atomes magnétogènes se traduira pour certains composés par l’apparition d’un ordre magnétique : ferromagnétisme, ferrimagnétisme, antiferromagnétisme, hélimagnétisme. Cette possibilité est très utilisée pratiquement. Par exemple, l’oxyde EuO (oxyde de Eu2+ équivalant électroniquement à Gd3+) est ferromagnétique (moments magnétiques alignés parallèlement), de même certains alliages comme Gd3Al2, Dy3Al2, etc., et surtout les alliages terres rares-cobalt du type Ln2Co17 et LnCo5 qui ont en outre des moments de saturation importants et des points de Curie très hauts (1 000 0C). Les substances ferrimagnétiques sont constituées de deux ou plusieurs sous-réseaux alignés de façon anti-parallèle avec des aimantations inégales en valeur absolue (théorie de Néel). C’est le cas des ferrites de lanthanides LnFe3 (pérovskites) et Ln3Fe512 (grenats) qui sont utilisées pour fabriquer des semiconducteurs.5. Propriétés chimiquesChimie en solutionLa chimie des lanthanides en solution est caractérisée par la prédominance quasi exclusive de la valence + 3 (la seule exception est l’europium, Eu2+), et par des réactions chimiques analogues tout au long de la série. Les ions en solution sont coordonnés par neuf molécules d’eau formant le complexe [Ln(H2O)9]+3. Certains sels (chlorures, nitrates) sont très solubles, d’autres très insolubles, notamment les oxalates (ceux-là, même en milieu acide: réaction de caractérisation et séparation des lanthanides en chimie analytique), les carbonates, les hydroxydes, les phosphates, les fluorures. La solubilité des composés peu solubles varie dans la série d’une façon irrégulière, par exemple la solubilité des hydroxydes décroît du lanthane au lutétium, elle croît pour d’autres sels (carbonates, sulfates), et, pour certains, elle passe par un maximum au niveau du gadolinium (glycollates). Les lanthanides en solution donnent de nombreux complexes avec les diacides organiques, et cela est utilisé pour les séparations. Les potentiels d’électrode E0(Ln3+aq /Ln) sont très négatifs, variant de – 2,52 V pour le lanthane à – 2,25 V pour le lutétium. Eu2+ en solution est analogue aux éléments alcalino-terreux et présente des réactions très voisines de celles de Sr2+.Chimie à l’état solideLa chimie de l’état solide des lanthanides est beaucoup moins monotone que la chimie en solution. Les structures cristallines sont souvent, mais pas toujours, identiques pour un composé donné, tout au long de la série. En général, dans ce cas, les paramètres du réseau décroissent de La à Lu, en raison de la contraction lanthanidique . En effet, à mesure que l’on ajoute des électrons f à l’atome, le rayon ionique de Ln3+ diminue. Il doit d’ailleurs être défini par rapport à un atome donné, car il est relativement variable: on obtient des valeurs différentes, par exemple, suivant que l’on part de la série des oxydes ou de celle des chlorures. Pour les oxydes, en prenant comme rayon ionique pour l’oxygène r = 0,138 nm, on obtient, d’après les distances Ln-O, des rayons qui varient de 0,102 nm pour le lanthane à 0,083 nm pour le lutétium.C’est dans l’état solide que l’on observe le plus facilement les autres valences des lanthanides. La valence +4 est obtenue pour le cérium, le praséodyme et le terbium dans les oxydes notamment, quelquefois pour d’autres éléments dans les fluorures complexes. La valence +2 est observée d’une façon très générale dans les systèmes métaux-halogénures et pour l’europium dans les oxydes. Les valences +2 sont également obtenues pour Eu dans certains sels (sulfates, carbonates).Les oxydes sont des composés très réfractaires qui fondent vers 2 200 0C. Ils possèdent trois structures cristallines principales, A hexagonale, B monoclinique et C cubique. Les structures A et B sont des phases de haute température, les températures de transformation montent dans la série. Les structures de ces oxydes, surtout A et B, n’ont pas d’équivalent pour d’autres éléments. En effet, elles reposent sur une coordination tétraédrique de l’oxygène par des atomes de terres rares. Les tétraèdres OLn4 ainsi définis sont joints par leurs arêtes et forment dans les structures A et B un véritable ion complexe à symétrie ternaire (LnO)nn +. Ces oxydes doivent donc se formuler (LnO)2O. La forme C-Ln23 est un assemblage tridimensionnel de tétraèdres OLn4 joints par quatre de leurs arêtes sur six. Le cation (LnO)nn + se rencontre aussi dans de très nombreux oxysels: oxychlorures (LnO)Cl, oxycarbonates (LnO)2C3, oxysulfures (LnO)2S, etc. L’arrangement des tétraèdres peut être aussi de symétrie quaternaire (LnOCl). Ces oxysels sont complètement insolubles, assez inertes chimiquement et généralement réfractaires, ils sont à la base de la variété observée dans la chimie de l’état solide par rapport à la chimie en solution. Ils forment aussi les meilleures matrices pour l’exploitation des propriétés optiques des ions Ln3+, du fait de l’influence de la liaison partiellement covalente Ln-O sur l’intensité des transitions.Les oxydes non stœchiométriques Cex, Prx et Tbx dus au mélange des valences +4 et +3 sont des exemples classiques de composés non stœchiométriques. Ils présentent de nombreuses phases intermédiaires, notamment pour des valeurs de x égales à 1,714, 1,778, 1,800, 1,833.Les atomes de terres rares entrent aussi dans la composition de nombreux oxydes mixtes avec d’autres éléments. Là encore, ils ont souvent tendance à coordonner tétraédriquement les atomes d’oxygène, par exemple dans les pyrochlores.Les chlorures, bromures et iodures anhydres sont extrêmement sensibles à l’humidité et ne peuvent être manipulés qu’en boîte à gants. On rencontre deux formes cristallines dans la série, une forme 見 type UCl3 (coordination 9 de la terre rare), et une forme 廓 type AlCl3 (coordination 6); la forme 見 au début, la forme 廓 à la fin de la série. Certains de ces halogénures ont aussi une structure du type PuBr3. Les points de fusion (600 à 900 0C) ont un minimum vers le milieu de la série. Les fluorures, beaucoup moins sensibles à l’humidité, sont plus réfractaires (point de fusion 1 500 à 1 200 0C, décroissant dans la série) et ont aussi deux structures La3 (hexagonale) au début de la série, Y3 (orthorhombique) après le gadolinium. Les chlorures fondus au contact du métal permettent d’obtenir pour presque tous les éléments les chlorures divalents très sensibles à l’oxydation.Les hydrures, borures, carbures, nitrures et sulfures sont des éléments d’un caractère beaucoup moins «ionique». Les hydrures LnHx sont non stœchiométriques, les borures ont pour formules LnB4 et LnB6, les carbures Ln3C, Ln2C3, les nitrures LnN, les sulfures Ln2S3 et LnS2. Ces substances présentent probablement des liaisons métaux-métaux, elles sont généralement d’une couleur très sombre et sont quelquefois des semiconducteurs ou même des conducteurs métalliques. Les lanthanides, dans ces composés, ont un comportement assez voisin de celui des éléments à électrons d , peut-être à cause de la participation d’électrons 5d aux liaisons.Les composés d’anions oxygénés sont très nombreux. Les hydroxydes ont une structure hexagonale où la terre rare est coordonnée par neuf ions OH-, les carbonates Ln2(CO3)3.n H2O (n = 8 au début de la série, 2 à 3 ensuite) et les nitrates Ln2(NO3)36H2O ont des structures caractérisées par une haute coordination de l’atome de terre rare, 10, 11 voire 12. Il en est probablement de même dans les autres sels hydratés, sulfates Ln2(SO4)3.8H2O, chlorures LnCl3.n H2O, n = 6 ou 7, etc., et les nombreux sels mixtes hydratés comportant d’autres cations.Les terres rares forment des complexes organiques assez nombreux, les plus intéressants étant les chélates de 廓-dicétones.La chimie de l’état solide est donc caractérisée par une grande variété: composés contenant un groupe à liaison partiellement covalente (LnO)nn +, composés à liaison métal-métal, sels beaucoup plus ioniques à haute coordination et composés de valence inhabituelle. Ces différences se voient dans la position des raies du spectre d’absorption de Ln3+, les raies sont déplacées vers le rouge pour les composés les plus covalents (oxydes, oxysels): c’est l’effet néphélauxétique.À haute température, il existe en outre des molécules LnO qui sont stables jusqu’à 6 000 K. On rencontre ces molécules dans certaines étoiles.MétallurgieLes métaux des terres rares sont préparés industriellement par la réaction du calcium avec le fluorure ou le chlorure anhydre à haute température et sous atmosphère inerte dans un creuset de tantale ou de tungstène. Les métaux ainsi obtenus sont en général assez purs au point de vue des éléments métalliques (Ca, Fe, Al, etc.) mais peuvent contenir des quantités notables d’impuretés non métalliques: hydrogène, oxygène, azote et carbone. Les métaux en couches minces sont de puissants getters , notamment pour l’hydrogène. Pour des expériences de recherche, il est nécessaire de purifier ces métaux, selon le cas, par refusion sous vide, sublimation, fusion de zone, ou électro-transport des impuretés à l’état solide.Les structures cristallines que présentent les métaux des terres rares sont très nombreuses. Leur description peut se faire au moyen des notations utilisées pour les matériaux polytypiques. Le tableau 3 donne un relevé des structures en fonction de la température, ainsi que le point de fusion et la pression de vapeur au point de fusion.Les électrons 4f demeurent localisés dans les lanthanides métalliques, les propriétés magnétiques sont donc celles des ions trivalents à l’exception de Eu et Yb, qui correspondent à des ions divalents, et du cérium qui est un cas de valence variable (entre 3 et 4). Ce sont surtout les lanthanides du groupe yttrique les plus lourds qui donnent lieu à des phénomènes d’ordre magnétique. Gd est ferromagnétique en dessous de 293,2 K 練 Tb, Dy, Ho et Tm ont deux structures magnétiques différentes dont l’une est simplement ferromagnétique et l’autre beaucoup plus complexe, les spins formant des hélices. Er possède trois structures magnétiques. Les complications sont dues au fait que l’axe de facile aimantation varie dans le cristal en fonction de la température.Ce, Y, Sc, Lu et surtout La sont supraconducteurs à haute pression. Pour La, la température critique est de 4,87 K à pression ordinaire et croît rapidement avec la pression (13 K à 200 kbar). Cependant, l’une des découvertes les plus remarquables est la mise en évidence de composés pour lesquels on observe simultanément un ordre magnétique et le phénomène de supraconductivité. C’est en particulier le cas pour les phases de Chevrel , GdMo6S8, TbMo6S8, qui sont à la fois supraconductrices et antiferromagnétiques à une température en moyenne inférieure à 2 K. Le composé ErRh4B4 présente une transition curieuse d’un état supraconducteur à un état ferromagnétique à 0,9 K.Les alliages formés par les métaux de terres rares sont très nombreux. On retiendra surtout les alliages avec le cobalt du type LnCo5 et les alliages ternaires avec le fer et le bore qui ont d’importantes applications comme aimants permanents. Les alliages amorphes formés avec les éléments du groupe 3d présentent quelquefois, malgré l’absence de structure cristallographique, de curieux phénomènes d’ordre magnétique: les moments pointent dans des directions déterminées par un cône ou une demi-sphère. On peut former dans ces matériaux (GdCo, GdFe, TbFe, etc.) des domaines magnétiques, par exemple sous forme de bulles, ils pourraient par conséquent servir de support à des informations lues ou écrites par des techniques laser thermomagnétiques.6. UtilisationsL’industrie des terres rares date de la fin du XIXe siècle, et elle a d’abord consommé essentiellement, soit des mélanges provenant de la monazite après extraction du thorium, soit des composés d’éléments faciles à séparer comme le cérium et le lanthane. Ce n’est que beaucoup plus tard, vers le milieu du XXe siècle, que se sont développées les utilisations basées sur les propriétés spécifiques de certains lanthanides (propriétés électroniques et spectrales en particulier).ComposésLes industries du verre et de la céramique comptent parmi les principales consommatrices de lanthanides. Des tonnages très importants d’oxydes de terres cériques, et plus spécialement d’oxyde de cérium, sont vendus sous forme de poudres pour polissage du verre (glaces, miroirs, prismes, lentilles et verres correcteurs d’optique médicale). Mais les oxydes de lanthanides interviennent aussi directement dans la fabrication de certains verres spéciaux: verre au lanthane à haut indice de réflexion et faible dispersion, verres de contraste au didyme (mélange de néodyme et praséodyme), verres protecteurs au néodyme absorbant les radiations jaunes des lampes au sodium, verres filtrants au cérium absorbant l’ultraviolet (verres de Crooks); l’oxyde de cérium, par son pouvoir oxydant, est aussi un excellent décolorant et stabilisant, il permet de préparer des verres de grande clarté et résistant aux radiations énergétiques (rayons X, rayons 塚). On emploie également les oxydes de lanthanides pour la verrerie d’art, en exploitant la couleur propre de leurs ions, soit pour compenser des colorations parasites (l’oxyde de néodyme est beaucoup employé dans ce but), soit pour obtenir des verres colorés (violet, rose ou pourpre avec le néodyme, vert avec le praséodyme, doré avec le cérium associé au titane).L’oxyde de cérium a été également beaucoup utilisé comme opacifiant pour les émaux, mais il est concurrencé par l’oxyde de titane, moins cher. L’oxyde de praséodyme sert à préparer un émail à base de silicate de zirconium d’un jaune intense, très apprécié pour la coloration de la porcelaine, et d’autres pigments contenant des terres cériques ont été mis au point récemment. L’emploi des oxydes de lanthanides a pris surtout de l’importance dans le domaine des céramiques industrielles: émaux et revêtements réfractaires, bougies d’allumage, et surtout céramiques diélectriques contenant par exemple du titanate ou du stannate de lanthane, à faible coefficient de température, utilisées dans les circuits résonnants (tels les oscillateurs de missiles). Les oxydes de terres rares entrent aussi, avec ceux de zirconium et de thorium, dans la composition d’éléments chauffants, conducteurs à haute température et résistant aux oxydants.L’industrie des catalyseurs a permis également aux lanthanides un débouché considérable, avec la mise au point de catalyseurs à base de terres rares pour le cracking du pétrole; ces catalyseurs améliorent le rendement en fractions légères du cracking et sont peu coûteux, car on les fabrique à partir de zéolites (alumino-silicates) et de mélanges de terres cériques (La, Nd, Pr). Cette application a pris aux États-Unis un développement très rapide et consomme, en tonnage, plus de la moitié de la production américaine d’oxydes de lanthanides; en Europe, par contre, ces catalyseurs sont beaucoup moins employés. Des catalyseurs à base de terres rares ont été brevetés pour de nombreuses autres réactions: polymérisations, isomérisations, hydrogénations et déshydrogénations, etc. Signalons, en particulier, l’oxydation catalytique de l’acide chlorhydrique pour l’obtention du chlore (ancien procédé Deacon) et l’utilisation de l’oxyde de cérium dans les moteurs d’automobile pour améliorer la combustion de l’essence.Une utilisation d’avenir est celle du cobaltite de lanthane comme catalyseur de réduction de l’oxygène dans les piles à combustibles des véhicules électriques.Les industries de l’éclairage sont probablement le domaine où l’utilisation des terres rares est le plus solidement implantée et offre les meilleures perspectives de développement, grâce aux propriétés spectrales spécifiques des lanthanides. Les propriétés émissives de l’oxyde de cérium, additionné en petites quantités à l’oxyde de thorium, ont fait le succès des «manchons Auer» d’éclairage au gaz; les «bâtonnets Nernst» sont toujours utilisés, comme sources de lumière très stables riches en infrarouges. Les oxydes et fluorures de lanthanides entrent depuis longtemps dans la fabrication des charbons à arc électrique intensif pour projecteurs; cette application consomme actuellement de 10 à 15 p. 100 de la production mondiale de lanthanides, principalement pour l’équipement des studios et salles de projection de cinéma, les équipements militaires, etc. Mais une application nouvelle a pris en peu de temps une importance considérable: c’est l’utilisation des lanthanides, et tout particulièrement de l’europium, dans les «phosphors», c’est-à-dire dans les matériaux luminescents employés dans les lampes d’éclairage. Dans ces substances, le lanthanide émetteur est introduit en petites quantités dans une «matrice» convenable, et l’émission lumineuse est obtenue par excitation à l’aide de radiations ultraviolettes (photoluminescence). Suivant l’élément de lanthanide choisi comme activateur, l’émission lumineuse est localisée dans le proche ultraviolet (avec le gadolinium, le cérium), le visible (rouge avec l’europium, orange avec le samarium, vert avec le terbium, jaune avec le dysprosium, bleu avec le thulium), ou le proche infrarouge (avec le néodyme). Cette propriété est utilisée pour améliorer la répartition spectrale des tubes fluorescents, et surtout dans les lampes à vapeur de mercure dans lesquelles un «phosphor» à base de terres rares, le vanadate d’yttrium activé à l’europium, transforme l’émission ultraviolette du mercure en lumière visible; ces lampes, qui donnent une lumière comparable à la lumière du jour, sont de plus en plus utilisées dans l’éclairage public. L’utilisation du vanadate d’yttrium activé à l’europium en cathodoluminescence (excitation par les électrons), pour les écrans de télévision en couleurs, a littéralement bouleversé l’industrie des terres rares dans les années soixante et, bien que le vanadate ait été supplanté par d’autres matrices (oxysulfures ou oxydes d’yttrium et de gadolinium), l’intérêt pour les propriétés spectrales de l’europium n’a pas diminué. Ce sont ces mêmes propriétés émissives qui sont exploitées, avec le néodyme, pour produire la lumière cohérente des lasers (verres ou monocristaux de grenat d’yttrium), et, avec divers lanthanides du groupe yttrique: Ho, Er, Yb, pour la construction de dispositifs transformant par des mécanismes de transfert d’énergie, les rayons infrarouges en lumière visible (compteurs quantiques).MétauxLes utilisations des lanthanides sous forme métallique consomment de 8 à 10 p. 100 de leur production globale. Elles font surtout appel à des mélanges, tels que le mischmétal (mélange des métaux cériques) utilisé en alliage avec 30 p. 100 de fer pour la fabrication des pierres à briquets. Cette industrie, qui date du début du siècle, est un monopole de producteurs de terres rares, et la production mondiale dépasse maintenant cinq cents tonnes par an, soit près de cinq milliards de pierres; l’application des propriétés pyrophoriques des alliages de lanthanides pour d’autres usages (pyrotechnie, signalisation, balles traceuses) reste modeste.Par contre, le mischmétal est de plus en plus utilisé en sidérurgie, comme désoxydant et désulfurant, pour la fabrication de fer ductile et d’aciers inoxydables à grains fins, pouvant être travaillés à chaud, et en fonderie, comme stabilisateur du carbone dans l’élaboration des fontes à graphite sphéroïdal: on peut dire qu’en Europe, pratiquement tous les alliages globulisants de la fonte contiennent du cérium additionné souvent sous forme d’un ferro-alliage spécial.Les métallurgistes utilisent aussi les métaux des lanthanides en alliages avec des métaux non ferreux, pour obtenir des propriétés améliorées: l’addition de mischmétal aux alliages nickel-chrome augmente leur résistance à la corrosion à chaud, et elle intervient dans la fabrication de presque toutes les qualités de fils électriques pour éléments chauffants: le céralumin (alliage aluminium-cérium) était utilisé dans la construction de pièces de moteurs d’avions dès la Première Guerre mondiale, et l’avènement de l’aviation supersonique et des fusées a beaucoup accru l’intérêt porté aux alliages légers améliorés aux terres rares: on observe une tendance à l’utilisation de métaux séparés de préférence aux mélanges, par exemple les alliages de magnésium-néodyme sont réputés pour leur excellente tenue mécanique à chaud.D’autre part, les propriétés magnétiques des métaux des lanthanides, qui n’avaient pas trouvé d’applications industrielles notables, sont passées au premier plan récemment avec la découverte des alliages de cobalt-lanthanides, de formule LnCo5 ou Ln2Co17, qui sont des aimants permanents plus puissants que tous ceux connus jusqu’à présent. On a fabriqué par exemple par frittage des aimants de samarium-cobalt (SmCo5) ayant des caractéristiques au moins deux fois plus élevées que les aimants de cobalt-platine industriels, et beaucoup moins chers que ces derniers; les performances théoriques de certains alliages cobalt-lanthanides (en particulier PrCo5) sont encore supérieures aux valeurs actuellement atteintes. Par ailleurs, la mise au point d’alliages ternaires Ln-Co-Fe ou Ln-Co-Cu préparés par fusion augmente encore l’intérêt de ces nouveaux aimants, utilisables non seulement pour les tubes micro-ondes et les autres dispositifs électroniques, mais également pour la construction des moteurs électriques.L’utilisation des lanthanides dans les technologies de pointe, magnétisme, électronique, sciences nucléaires, offre des perspectives d’avenir très variées:– Parmi les applications en cours de développement, on peut citer les masers et lasers (monocristaux de grenat d’yttrium-aluminium dopés au néodyme) utilisés en optique, télémétrie et télécommunications, les grenats ferrimagnétiques d’yttrium et de gadolinium, employés dans les dispositifs électroniques utilisant les micro-ondes, les orthoferrites (LnFeO3), les monochalcogénures d’europium (EuO, EuS, EuSe, EuTe) dont les propriétés magnéto-optiques sont exploitables pour des fonctions de mémoire ou de transmission dans les ordinateurs, les borures (de lanthane, en particulier) qui servent à fabriquer des cathodes émissives pour canons à électrons et tubes électroniques divers.– L’industrie nucléaire, d’autre part, utilise les très fortes sections efficaces d’absorption des neutrons (du samarium, du gadolinium, du dysprosium ou de l’europium) pour la fabrication de barres de contrôle et de réglage dans les réacteurs nucléaires, ou dans les revêtements de protection contre les radiations. Enfin, les radio-éléments artificiels des lanthanides (produits de fission ou obtenus par irradiation) offrent un large choix d’utilisations futures, basées sur leurs radiations: sources portatives de rayons X ou rayons 塚 pour radiographie et radioscopie, usages thérapeutiques, composés radio-luminescents activés au prométhium, micro-générateurs d’électricité par conversion d’énergie radio-voltaïque, etc.Toutes ces applications nouvelles, faisant appel à la spécificité des propriétés des lanthanides, permettent de penser que ces éléments prendront dans l’avenir une importance industrielle croissante, et expliquent que les spécialistes les considèrent comme les «matériaux du XXIe siècle».
Encyclopédie Universelle. 2012.